Urea cycle
The urea cycle detoxifies ammonia, a toxic byproduct of amino acid catabolism, by converting it into excretable urea in hepatocytes, consuming energy equivalents of four high-energy phosphate bonds per urea molecule produced.
Cycle Overview
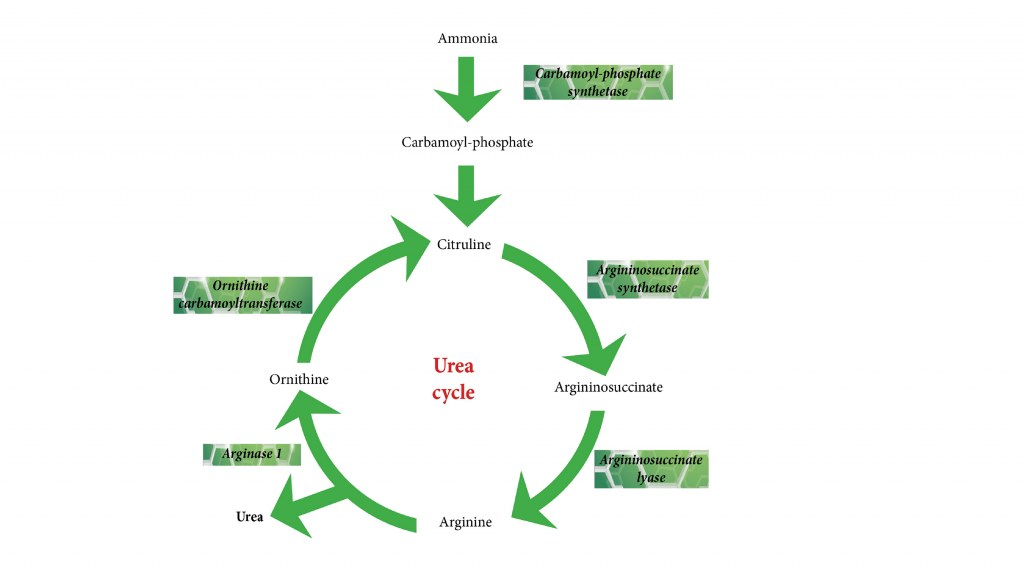
The urea cycle spans mitochondria (steps 1-2) and cytosol (steps 3-5) of liver cells, incorporating two nitrogen atoms—one from ammonia (NH₄⁺), the other from aspartate—into urea (H₂N-CO-NH₂). Ornithine shuttles between compartments as a carrier, while fumarate from step 4 links to the citric acid cycle via aspartate-argininosuccinate shunt.
Detailed Enzymatic Steps
Step 1 (Mitochondria): NH₄⁺ + HCO₃⁻ + 2 ATP → carbamoyl phosphate + 2 ADP + Pi + H⁺, catalyzed by carbamoyl phosphate synthetase I (CPS1), activated allosterically by N-acetylglutamate (NAG).
Step 2 (Mitochondria): Carbamoyl phosphate + ornithine → citrulline + Pi, via ornithine transcarbamylase (OTC); citrulline exits via transporter.
Step 3 (Cytosol): Citrulline + aspartate + ATP → argininosuccinate + AMP + PPi, by argininosuccinate synthetase (ASS1), rate-influenced by substrate availability.
Step 4 (Cytosol): Argininosuccinate → arginine + fumarate, cleaved by argininosuccinate lyase (ASL); fumarate converts to malate and thence aspartate.
Step 5 (Cytosol): Arginine + H₂O → urea + ornithine, hydrolyzed by arginase (ARG1), regenerating ornithine for mitochondrial re-entry.
|
|
Regulation and Disorders
CPS1, the rate-limiting enzyme, responds to NAG (induced by high protein intake via NAGS), with feedback from ornithine/citrulline. Defects (e.g., OTC deficiency) cause hyperammonemia, treated by dialysis, benzoate, or liver transplant.
Overall: 2 NH₃ + CO₂ + 3 ATP + aspartate → urea + fumarate + 2 ADP + AMP + PPi + 2 Pi.